How to Set Up a Pharmaceutical Data Room for Licensing and M&A Deals
A pharmaceutical data room is a secure virtual workspace used during drug licensing deals, pharma M&A, and partnership transactions to share clinical trial data, regulatory filings, manufacturing documentation, and IP portfolios. This guide covers how to organize one for large pharma transactions, what documents belong in each section, and how to manage access across deal stages.

Why Pharma Deals Require Specialized Data Rooms
Pharmaceutical transactions are document-heavy by nature. A typical pharma licensing deal data room contains between 5,000 and 15,000 documents spanning clinical results, manufacturing records, patent filings, and commercial projections. That volume alone makes general-purpose file sharing tools impractical.
The stakes are enormous. Global pharma M&A deal value reached roughly $250 billion across 516 deals in 2025, with average deal sizes climbing further in early 2026. Licensing activity hit records too, with 516 out-licensing deals signed in 2025 alone. Each of these transactions depends on a data room that buyers, licensees, and their advisors can navigate efficiently.
What separates a pharma data room from a generic virtual data room comes down to three factors:
- Regulatory complexity: FDA and EMA submissions, clinical hold letters, chemistry/manufacturing/controls (CMC) packages, and post-market commitments create document categories that don't exist in other industries
- Multi-party access requirements: A single licensing deal may involve the licensor, licensee, their respective legal counsel, regulatory consultants, CMO auditors, and financial advisors, each needing different document access
- Staged disclosure: Sensitive manufacturing processes, patient-level clinical data, and pre-submission regulatory strategies are shared progressively as the deal advances through diligence phases
The companies that run clean data rooms close faster. When a pharma business development team opens your room and finds a logical folder structure with clear naming conventions, it signals operational maturity. When they find 3,000 unlabeled PDFs in a flat folder, they start questioning what else is disorganized.
Helpful references: Fastio Workspaces, Fastio Collaboration, and Fastio AI.
What to check before scaling pharmaceutical data room
The right folder structure lets reviewers find what they need without asking. For pharma licensing and M&A deals, organize your room around six core sections.
1. Regulatory Filings and Correspondence
This is usually the first section reviewers open. Include:
- IND/IMPD applications and amendments
- FDA/EMA meeting minutes and official correspondence
- Clinical hold letters and responses
- Marketing authorization applications (NDA, BLA, MAA)
- Annual reports and periodic safety update reports (PSURs)
- Pharmacovigilance plans and risk management documentation
- Country-specific regulatory submissions for markets where the drug is filed
Organize by regulatory authority first, then chronologically within each authority. Reviewers expect to trace the regulatory history of each product candidate from initial filing through current status.
2. Clinical Trial Data Structure clinical data by trial phase, then by study identifier:
- Clinical study reports (CSRs) for completed trials
- Trial protocols and amendments
- Statistical analysis plans (SAPs)
- Interim analysis reports
- Data Safety Monitoring Board (DSMB) meeting minutes
- Informed consent forms (ICFs)
- Trial Master File (TMF) index following the DIA TMF Reference Model
- Site-level enrollment and performance data
- Adverse event summaries and safety narratives
For Phase III assets and commercial products, buyers expect the full CSR package. For earlier-stage compounds, protocols and interim data may suffice, but be transparent about what is and isn't available.
3. Chemistry, Manufacturing, and Controls (CMC)
CMC documentation is where many deals slow down. Reviewers are evaluating whether the drug can be manufactured reliably at scale, and gaps here raise serious red flags. Include:
- Drug substance and drug product specifications
- Analytical method validation reports
- Stability study data (accelerated and long-term)
- Process validation reports
- Batch records for clinical and registration batches
- GMP certificates for manufacturing sites
- CMO/CDMO contracts and quality agreements
- Supply chain documentation and raw material sourcing
- Comparability studies (if manufacturing changes occurred)
Follow ALCOA+ principles for all CMC data: attributable, legible, contemporaneous, original, accurate, complete, consistent, enduring, and available. Reviewers from pharma quality teams will check.
4. Intellectual Property Portfolio
Patent documentation should be organized by patent family:
- Granted patents with filing and expiration dates
- Pending applications and prosecution history
- Freedom-to-operate (FTO) opinions
- Patent term extensions and supplementary protection certificates
- Trade secret inventories (redacted as appropriate for deal stage)
- Licensing agreements affecting IP rights
- Patent landscape analyses for the therapeutic area
5. Commercial and Financial Projections
- Revenue forecasts by market and indication
- Pricing and reimbursement analysis by country
- Market research reports and competitive intelligence
- Sales force structure and commercial launch plans
- Payer access strategies
- Historical sales data (for marketed products)
- Royalty and milestone payment schedules from existing agreements
6. Legal and Corporate
- Material contracts (licensing agreements, co-development agreements, co-promotion agreements)
- Litigation history and pending legal matters
- Corporate structure and subsidiary information
- Key employee agreements and non-compete clauses
- Insurance policies
- Environmental and safety compliance records for manufacturing facilities
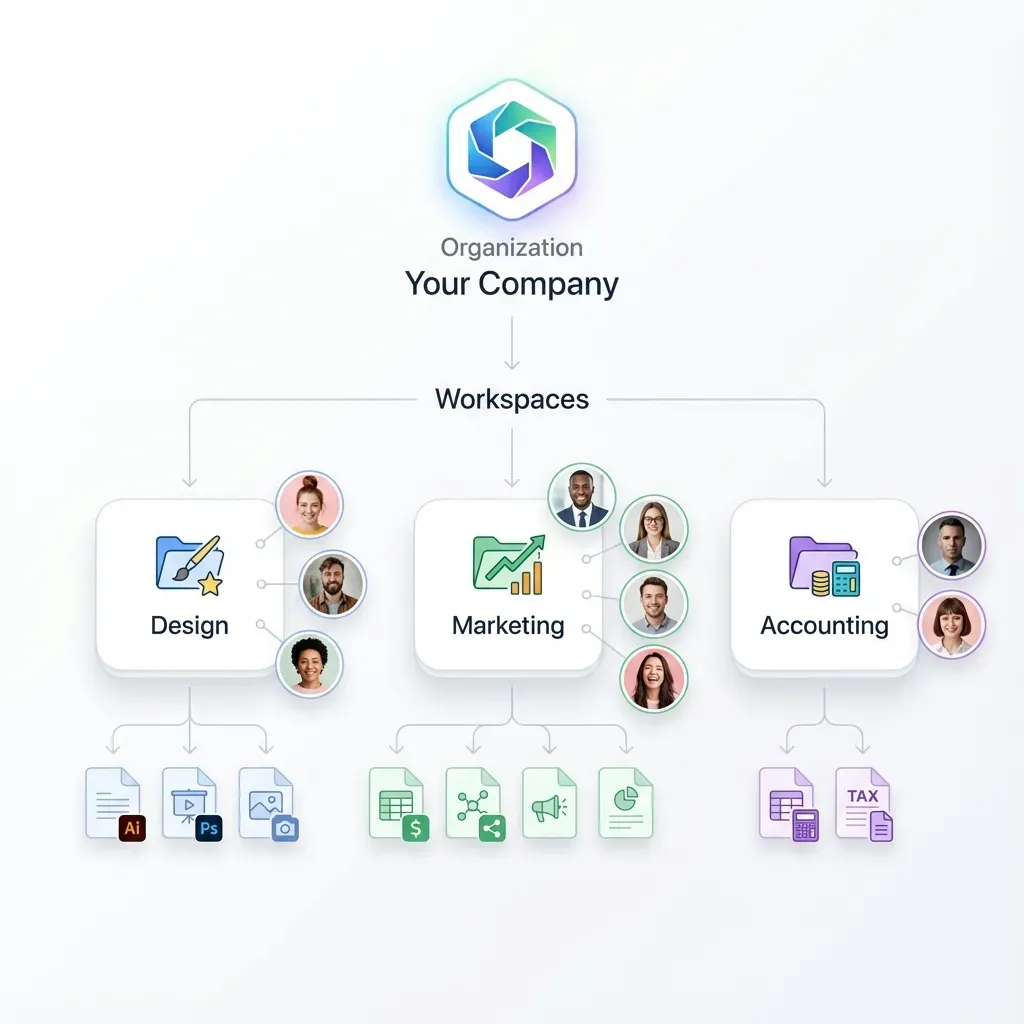
Managing Access and Permissions Across Deal Stages
Pharma deals don't share everything on day one. Access should expand as the deal moves through stages, and different advisor groups need different views of the room.
Phase 1: Initial Interest (Limited Access)
When a potential licensee or acquirer first enters the room, they typically see:
- Executive summary and product overview
- Published clinical data and regulatory milestones
- Patent summaries (not full prosecution files)
- High-level commercial projections
This is the "teaser" stage. You want to give enough information for the counterparty to decide whether to proceed to full diligence, without exposing sensitive manufacturing data or unpublished clinical results.
Phase 2: Full Due Diligence (Expanded Access)
Once an NDA or confidentiality agreement is in place, open additional folders:
- Complete clinical study reports
- CMC documentation and manufacturing records
- Detailed IP prosecution files and FTO opinions
- Financial models with assumptions
- Regulatory correspondence and meeting minutes
At this stage, different advisor groups should see different slices. Regulatory consultants need clinical and regulatory folders but not financial models. Financial advisors need projections and contracts but not raw CMC data.
Phase 3: Confirmatory Diligence (Full Access)
In the final stage before signing, the remaining restricted documents open:
- Patient-level clinical data (de-identified per strict security requirements/privacy requirements requirements)
- Detailed manufacturing process descriptions and trade secrets
- Internal quality audit reports
- Board minutes and governance documents
Granular permissions matter here. You need folder-level and file-level access controls, not just room-level. Fastio supports permissions at the organization, workspace, folder, and individual file level, which maps directly to this staged disclosure model. Each advisor group gets a permission set matching their role.
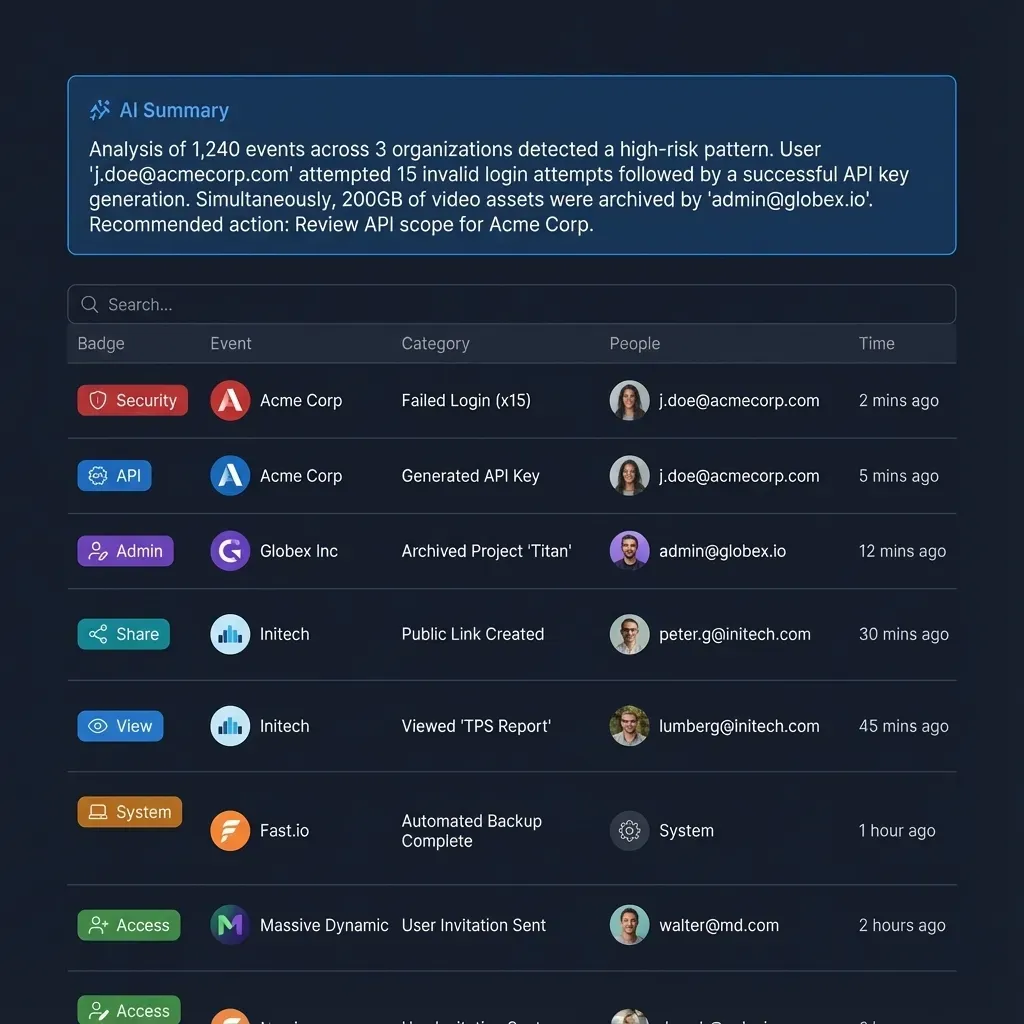
Audit trails are equally important. Every document view, download, and print should be logged with timestamps and user identity. When deal teams from both sides are accessing thousands of documents over weeks or months, a clear activity record protects everyone. Fastio's audit trail captures file operations, membership changes, and activity across workspaces, giving deal administrators visibility into exactly who accessed what and when.


Build Your Pharma Data Room Today
Set up a secure, organized data room for your next licensing deal or acquisition. Start with generous storage, granular permissions, and built-in document intelligence. Built for pharmaceutical data room workflows.
CMO Audits and Manufacturing Due Diligence
Contract manufacturing organization (CMO) audits are a critical part of pharma M&A that biotech-focused data room guides rarely cover in depth. When a buyer acquires a pharmaceutical company or licenses a product, they inherit the manufacturing relationships. The data room needs to prove those relationships are solid.
What CMO Audit Reviewers Look For Manufacturing due diligence teams focus on:
- GMP compliance history: Inspection reports from FDA, EMA, and other regulatory authorities for each manufacturing site. Any Form 483 observations, warning letters, or consent decrees should be disclosed with remediation documentation.
- Quality agreement completeness: The quality agreement between sponsor and CMO defines responsibilities for release testing, stability programs, deviation handling, and change control. Gaps here are deal-breakers.
- Technology transfer readiness: If the buyer plans to move manufacturing, they need process descriptions detailed enough to replicate at a new site. This includes process parameters, critical quality attributes (CQAs), and critical process parameters (CPPs).
- Supply continuity risk: Dual sourcing arrangements, safety stock levels, raw material qualification status, and lead times for critical starting materials.
- Batch failure rates and deviation trends: Not just current performance, but trends over the last 12 to 24 months. Rising deviation rates or out-of-specification (OOS) results signal manufacturing process drift.
Organizing CMO Documentation in the Data Room
Create a subfolder per manufacturing site and CMO relationship. Within each:
- Current GMP certificate and regulatory inspection history
- Quality agreement (current version plus amendment history)
- Annual product quality reviews (APQRs)
- Process validation summary reports
- Stability data for batches produced at the site
- Change control log with impact assessments
- Vendor qualification records for critical raw materials
Many pharma companies make the mistake of keeping CMO documentation in the same folders as internal manufacturing data. Separate them. Buyers and their manufacturing consultants need to evaluate each CMO independently.
Regulatory Submission Workflows in the Data Room
For pharma licensing deals involving marketed products or late-stage candidates, the data room often serves double duty. It's not just a repository for completed regulatory filings. It also needs to support ongoing submission workflows during the deal process.
Common Scenarios
- Pre-approval deals: The licensor may be preparing an NDA or MAA while the data room is active. New sections of the Common Technical Document (CTD) need to be added as they're completed.
- Lifecycle management: Post-approval supplements, label expansions, and variations may be in progress during the transaction.
- Multi-market filings: A global licensing deal may involve simultaneous regulatory submissions in the US, EU, Japan, and other markets, each with different document requirements.
Version Control and Document Currency
Regulatory submissions evolve constantly. The data room needs to handle version control cleanly so reviewers always know they're looking at the current version. Fastio supports file versioning, so you can update a CMC section or clinical summary without breaking links or losing the revision history. Reviewers can see the current version while still being able to access earlier iterations if they need to trace changes.
CTD Module Organization
For deals involving regulatory submissions, consider organizing one section of the data room to mirror the CTD structure:
- Module 1: Administrative information and prescribing information (region-specific)
- Module 2: CTD summaries (quality overall summary, nonclinical overview, clinical overview)
- Module 3: Quality (drug substance, drug product, stability)
- Module 4: Nonclinical study reports
- Module 5: Clinical study reports
This mirrors the structure that regulatory reviewers already know, reducing the learning curve for anyone evaluating submission readiness.
Collaboration During Active Submissions
Some deals require real-time collaboration on regulatory documents. For example, a licensing partner may need to review and comment on draft labeling or a risk management plan before submission. Fastio's commenting system supports anchored feedback on specific document sections, pages, and selections, which keeps regulatory review discussions organized and traceable rather than scattered across email threads.
Setting Up Your Pharma Data Room
Getting the practical setup right saves weeks during the deal process. Here's how to approach it.
Choose the Right Platform
General-purpose cloud storage services like Google Drive or Dropbox work for basic file sharing, but pharma deals demand more. You need granular permissions, audit trails, versioning, and the ability to handle thousands of documents without performance issues.
Dedicated VDR providers like Intralinks, Datasite, and Firmex have long dominated the pharma deal market. They're proven but expensive, often charging per-page or per-user fees that add up quickly when you're sharing 10,000 documents with 50 advisors.
Fastio offers an alternative approach with workspace-based organization, folder and file-level permissions, audit trails, and built-in AI that indexes your documents for search and Q&A. The free plan includes 50 GB of storage, which is enough for many early-stage or mid-size deal rooms. For larger transactions, the usage-based credit model avoids the per-page pricing that makes traditional VDRs expensive. Intelligence Mode automatically indexes uploaded documents for semantic search, so reviewers can ask questions across the entire data room rather than manually searching through thousands of files.
Naming Conventions
Consistent file naming prevents confusion when dozens of people are navigating the room:
- Start with the document category:
REG_FDA_IND-123456_Annual-Report_2025.pdf - Include version numbers for living documents:
CMC_Drug-Product-Spec_v3.2.pdf - Use dates in ISO format:
CLI_CSR_Study-001_2025-06-15.pdf - Avoid spaces in filenames. Use hyphens or underscores.
Index Documents
Create a master document index as a spreadsheet in the root folder. Include:
- Document name and folder location
- Document date and version
- Confidentiality classification (public, confidential, highly restricted)
- Access tier (Phase 1, Phase 2, or Phase 3 access)
This index serves as the table of contents for your data room. Update it whenever documents are added or moved.
Prepare Before the Deal Starts
The best time to build a pharma data room is before you need one. Companies that maintain a "standing data room" with current regulatory filings, IP documentation, and CMC records can launch a deal process in days instead of weeks. Assign someone in regulatory affairs or business development to keep core documents current. When a deal opportunity arises, you copy the standing room into a deal-specific workspace and add transaction-specific materials.
Fastio's workspace model supports this approach naturally. Maintain a master workspace with your standing documents, then create deal-specific workspaces with the relevant subset. Branded shares let you present the room with your company identity, and auto-expiring access links ensure that access ends when the deal closes.
Frequently Asked Questions
What documents go in a pharmaceutical data room?
A pharmaceutical data room typically contains six categories of documents. Regulatory filings and correspondence (IND applications, FDA meeting minutes, marketing authorization applications). Clinical trial data (study reports, protocols, safety data). CMC and manufacturing documentation (process validation, stability data, GMP certificates, CMO contracts). Intellectual property (patents, FTO opinions, prosecution history). Commercial projections (revenue forecasts, pricing analysis, market research). Legal and corporate documents (material contracts, litigation history, corporate structure).
How do pharma companies structure data rooms for licensing deals?
Pharma licensing data rooms are organized around the six core sections listed above, with access staged across the deal timeline. Initial access includes executive summaries and published data. Full due diligence opens clinical study reports, CMC documentation, and IP files. Confirmatory diligence provides patient-level data and manufacturing trade secrets. Different advisor groups (regulatory, legal, financial, manufacturing) receive access only to the sections relevant to their review scope.
What is the difference between a biotech and pharma data room?
Biotech data rooms tend to focus on fundraising and early-stage partnering, typically containing fewer documents (500 to 2,000) organized around a single lead candidate. Pharmaceutical data rooms support larger, more complex transactions with 5,000 to 15,000 documents spanning multiple products, marketed assets, manufacturing networks, and global regulatory portfolios. Pharma rooms also require deeper CMO audit documentation, multi-market regulatory filings, and commercial performance data that biotech rooms rarely include.
How long does it take to set up a pharma data room?
Starting from scratch, building a comprehensive pharma data room takes 4 to 8 weeks. Most of that time goes into collecting CMC documentation, organizing regulatory correspondence, and preparing document indices. Companies that maintain a standing data room with current core documents can launch a deal-specific room in 3 to 5 days. The setup time also depends on how many products and manufacturing sites are involved.
What security features should a pharma data room have?
At minimum, a pharma data room needs granular access controls (folder and file level), comprehensive audit trails logging every view and download, version control for evolving documents, and strong authentication. Dynamic watermarking, download restrictions, and auto-expiring access links add additional protection. For clinical data containing patient information, the platform should support access controls strict enough to maintain strict security requirements and privacy requirements requirements for de-identified data handling.
How much does a pharmaceutical data room cost?
Traditional VDR providers charge between $15,000 and $100,000 per deal room, depending on volume, user count, and deal duration. Per-page pricing models become expensive quickly with 10,000-plus document rooms. Usage-based platforms like Fastio offer an alternative, starting with a free tier (50 GB storage, no credit card required) and scaling based on actual usage rather than document count or seat count.
Related Resources

Build Your Pharma Data Room Today
Set up a secure, organized data room for your next licensing deal or acquisition. Start with generous storage, granular permissions, and built-in document intelligence. Built for pharmaceutical data room workflows.